
Mucopolisacaridosis (MPS) VI, o síndrome Maroteaux-Lamy, es una enfermedad devastadora, progresiva y heterogénea con diversas patologías que afecta múltiples órganos. Es causada por una actividad deficiente de arilsulfatasa B (ASB), la enzima que cataboliza los glicosaminoglicanos (GAGs) dermatán sulfato y condroitín sulfato.1
A pesar de que existe un gran variabilidad de presentación fenotípica de MPS VI,1,2 los pacientes generalmente se caracterizan por tener una enfermedad de progresión rápida o lenta:
Independientemente del índice de progresión, la MPS VI sin tratamiento puede progresar con lo años y ocasionar lo siguientes 3:
MPS VI es un cuadro clínicamente heterogéneo en el que los pacientes pueden presentar una enfermedad marcada en el primer año de vida, o una enfermedad que progresa de manera más lenta con síntomas que aparecen a lo largo del tiempo. Sin embargo, es importante reconocer que la MPS VI manifiesta síntomas continuamente, por lo que no existen parámetros establecidos para estas categorías de progresión de la enfermedad.1,2
La MPS VI de progresión rápida, caracterizada en el estudio transversal realizado por Swiedler et como niveles de proteína GAG en orina (uGAG) por encima de 200 μg/mg,3 puede manifestarse en el primer año de vida con síntomas no específicos. Entre los 2 y 3 años se desarrollan los rasgos característicos, incluyendo los siguientes 1,2,5:
La MPS VI de progresión lenta, caracterizada como niveles uGAG menores o iguales a 200 μg/mg, puede no estar manifiesta, por lo que demora el diagnóstico hasta varios años después, no obstante, los pacientes desarrollan una morbidez significativa que puede ser una limitación y amenaza para la vida. Ya sea de progresión lenta o rápida, la MPS VI no suele causar deficiencias neurocognitivas, a pesar de que las limitaciones físicas pueden afectar el aprendizaje y el desarrollo de los pacientes.3
La tabla a continuación describe las complicaciones de MPS VI por sistema corporal. Las manifestaciones clínicas asociadas con MPS VI son heterogéneas, todos los pacientes experimentarán la progresión de la enfermedad.

Los pacientes con MPS VI expresan tasas de crecimiento significativamente menores que sus pares no afectados de la misma edad. El desvío en el crecimiento de los pacientes con MPS VI puede ser un signo de problemas secundarios y debe indicar una rápida investigación clínica de cuestiones como mala nutrición, anormalidades endocrinólogas o privación psicológica.

La MPS VI es un cuadro clínicamente heterogéneo con un espectro de fenotipos de aparición y tasas de progresión variables.2 La figura a continuación muestra la gran variabilidad entre un paciente con progresión rápida y un paciente con progresión lenta. El paciente con progresión rápida presenta muchos de los rasgos distintivos de MPS VI.
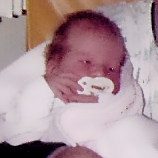
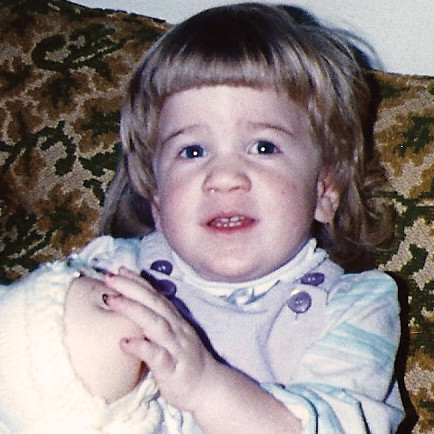


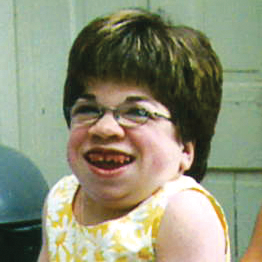
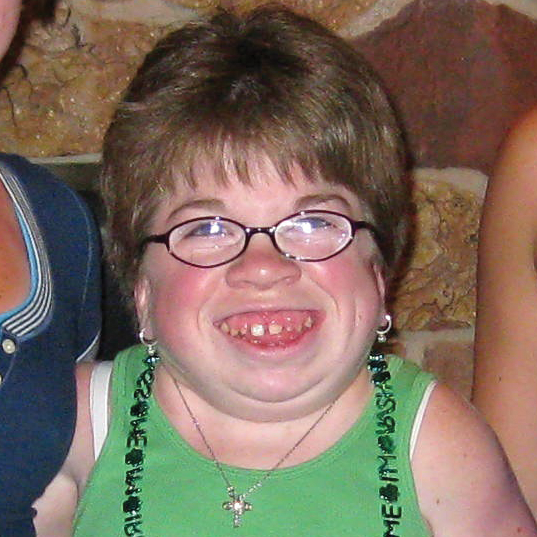
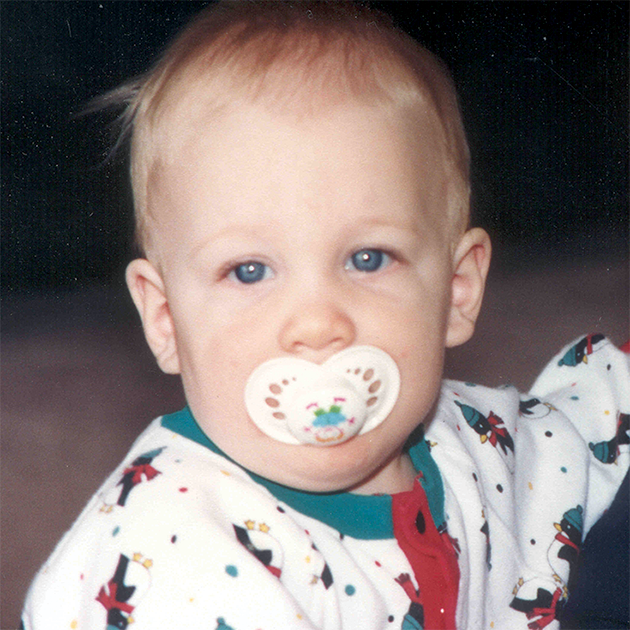

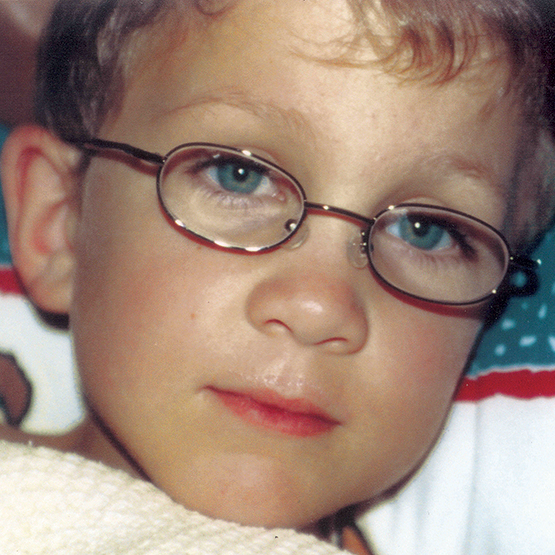



Diferenciación entre enfermedad de progresión lenta y rápida es aparente sólo en casos extremos. Sin tratamiento, la progresión de la enfermedad inhibirá el bienestar físico y funcional y resultará en una vida significativamente más corta.4
Independientemente del fenotipo, los individuos afectados progresan durante el curso de la enfermedad y finalmente presentan 4
Los pacientes con MPS VI normalmente fallecen a causa de infecciones, complicaciones secundarias a la cirugía, o enfermedad cardiopulmonar.4
MPS VI es un cuadro recesivo autosomal causado por una deficiencia de la enzima N-acetilgalactosamina 4-sulfatasa (también conocida como ASB). Esta la deficiencia resulta en la acumulación de GAG dermatán sulfato en los lisosomas de una gran variedad de tejidos13,14
Una mirada histológica sobre la patofisiología de la acumulación GAG en MPS VI

No se asoció ningún grupo étnico específico con un mayor riesgo de MPS VI; sin embargo, se han informado algunas frecuencias mayores de mutaciones específicas. Existe una mutación común (1533del23) encontrada en el 23% de alelos en pacientes brasileños con MPS VI. Además, un estudio demostró un alta prevalencia de MPS VI al nacimiento en la población turca que vive en Alemania, en comparación con la población alemana no turca.2
En la última década, el tratamiento de MPS VI evolucionó junto con el conocimiento de los clínicos acerca de la enfermedad, como por ejemplo acerca de su variabilidad fenotípica y progresión. Otros factores, como pacientes que viven hasta la edad adulta, destacan nuevas áreas de investigación, conocimiento y promesas para el tratamiento.
MPS VI afecta múltiples sistemas corporales, lo que hace que el tratamiento de un diverso espectro de manifestaciones de la enfermedad sea una parte importante del cuidado integral. La atención debe incluir el uso de dispositivos de adaptación o soporte, terapia física ocupacional, medicaciones en base a los síntomas, intervenciones quirúrgicas y tratamiento para proveer la enzima deficiente.4
Publicados el año en 2007, los “Management Guidelines for Mucopolysaccharidosis” (Lineamientos de Atención a Mucopolisacaridosis) establecen recomendaciones para todas las especialidades en la atención de pacientes con MPS VI. Recomiendan la terapia de reemplazo enzimático (TRE) como la opción de tratamiento a tener en cuenta para pacientes con MPS VI.4
Además de iniciar la TRE cuando corresponda, los lineamientos recomiendan estrategias de atención específicas para los síntomas asociados con la acumulación de GAG.4 La atención de por vida y el cuidado quirúrgico por parte de un equipo coordinado multidisciplinario son componentes críticos para optimizar los resultados en pacientes.16 Los pacientes con MPS VI requieren evaluaciones tempranas y regulares para evaluar la progresión de la enfermedad en todos los sistemas corporales y para detectar y tratar posibles complicaciones de la enfermedad.4 La tabla a continuación muestra el cronograma de evaluaciones para pacientes con MPS VI recomendado por los lineamientos del 2007.

Los pacientes con MPS VI a menudo requieren una intervención quirúrgica para tratar las complicaciones multisistémicas de la enfermedad. Este cuidado quirúrgico es complicado por la naturaleza de la enfermedad.16
Los pacientes con MPS VI sufren múltiples factores que pueden aumentar el riesgo y la necesidad de monitoreo de manera dramática, incluyendo los siguientes16:
Estos factores complican el cuidado quirúrgico y anestésico, requieren planeamiento y necesitan técnicas específicas de la enfermedad para obtener mejores resultados.17
Los procedimientos perioperatorios especializados durante la anestesia, tales como intubación y extubación, y el uso de una lista de verificación de monitoreo neurológico intraoperatorio, son esenciales para el éxito de toda intervención quirúrgica. Para obtener resultados duraderos y positivos es fundamental contar con un equipo quirúrgico integral formado por un especialistas en MPS VI.16,17
A medida que los pacientes con MPS VI alcanzan la edad adulta, su relación con el equipo médico se modifica. Para colaborar en esta transición, se necesitan planes individuales a fin de minimizar las interrupciones del tratamiento, extender el soporte más allá de la atención pediátrica y apoyo de los padres y asegurar que los pacientes adultos sepan como tratar la MPS VI.18
Estos planes de transición deben ser elaborados de acuerdo con las necesidades específicas d de cada paciente, de modo que aquellos que puedan ocuparse de su propio cuidado posean las herramientas necesarias y que aquellos que están más limitados cuenten con los cuidados y servicios necesarios. Los planes deben incluir una evaluación para determinar la capacidad del paciente para lograr sus metas definidas, así como también su conocimiento y capacidad para informar acerca de su estado de salud.18
References: 1. Thümler A, Miebach E, Lampe C, et al. Clinical characteristics of adults with slowly progressing mucopolysaccharidosis VI: a case series. J Inherit Metab Dis. 2012;35(6):1071-1079. doi:10.1007/s10545-012-9474-1. 2. Valayannopoulos V, Nicely H, Harmatz P, Turbeville S. Mucopolysaccharidosis VI. Orphanet J Rare Dis. 2010;5:5. doi:10.1186/1750-1172-5-5. 3. Swiedler SJ, Beck M, Bajbouj M, et al. Threshold effect of urinary glycosaminoglycans and the walk test as indicators of disease progression in a survey of subjects with mucopolysaccharidosis VI (Maroteaux–Lamy syndrome). Am J Med Genet A. 2005;134A(2):144-150. doi:10.1002/ajmg.a.30579. 4. Giugliani R, Harmatz P, Wraith JE. Management guidelines for mucopolysaccharidosis VI. Pediatrics. 2007;120:405-418. doi:10.1542/peds.2006-2184. 5. Muhlebach MS, Wooten W, Muenzer J. Respiratory manifestations in mucopolysaccharidoses. Paediatr Respir Rev. 2011;12(2):133-138. doi:10.1016/j.prrv.2010.10.005. 6. Lin H-Y, Chen M-R, Lin C-C, et al. Polysomnographic characteristics in patients with mucopolysaccharidoses. Pediatr Pulmonol. 2010;45(12):1205-1212. doi:10.1002/ppul.21309. 7. Kampmann C, Lampe C, Whybra-Trumpler C, et al. Mucopolysaccharidosis VI: cardiac involvement and the impact of enzyme replacement therapy. J Inherit Metab Dis. 2014;37(2):269-276. doi:10.1007/s10545-013-9649-4. 8. Willoughby CE, Ponzin D, Ferrari S, Lobo A, Landau K, Omidi Y. Anatomy and physiology of the human eye: effects of mucopolysaccharidoses disease on structure and function—a review. Clin Exper Ophthalmol. 2010;38(suppl 1):2-11. doi:10.1111/j.1442-9071.2010.02363.x. 9. Ganesh A, Bruwer Z, Al-Thihli K. An update on ocular involvement in mucopolysaccharidoses. Curr Opin Ophthalmol. 2013;24(5):379-388. doi:10.1097/ICU.0b013e3283644ea1. 10. Kantaputra PN, Kayserili H, Güven Y, et al. Oral manifestations of 17 patients affected with mucopolysaccharidosis type VI. J Inherit Metab Dis. 2014;37(2):263-268. doi:10.1007/s10545-013-9645-8. 11. Quartel A, Hendriksz CJ, Parini R, Graham S, Lin P, Harmatz P. Growth charts for individuals with mucopolysaccharidosis VI (Maroteaux-Lamy Syndrome). JIMD. 2015;18:1-11. doi:10.1007/8904_2014_333. 12. Data on file. BioMarin Pharmaceutical Inc. 13. NAGLAZYME [package insert]. Novato, CA: BioMarin Pharmaceutical Inc; 2013. 14. Giugliani R, Lampe C, Guffon N, et al. Natural history and galsulfase treatment in mucopolysaccharidosis VI (MPS VI, Maroteaux-Lamy syndrome)—10-year follow-up of patients who previously participated in an MPS VI Survey Study. Am J Med Genet A. 2014;164A(8):1953-1964. doi:10.1002/ajmg.a.36584. 15. Kakkis ED. Enzyme replacement therapy for the mucopolysaccharide storage disorders. Expert Opin Investig Drugs. 2002;11(5):675-685. 16. Walker R, Belani KG, Braunlin EA, et al. Anaesthesia and airway management in mucopolysaccharidosis. J Inherit Metab Dis. 2013;36(2):211-219. doi:10.1007/s10545-012-9563-1. 17. Vitale MG, Skaggs DL, Pace GI, et al. Delphi Consensus Report: Best practices in intraoperative neuromonitoring in spine deformity surgery: development of an intraoperative checklist to optimize response. Spine Deformity. 2014;2(5):333-339. doi:10.1016/j.jspd.2014.05.003. 18. American Academy of Pediatrics, American Academy of Family Physicians, American College of Physicians, Transitions Clinical Report Authoring Group, Cooley WC, Sagerman PJ. Supporting the health care transition from adolescence to adulthood in the medical home. Pediatrics. 2011;128(1):182-200. doi:10.1542/peds.2011-0969.


